
宏基因组学是指利用高通量测序等方法,对环境样本中的微生物群体进行基因测序,以获得微生物的基因功能、组成和丰度。宏基因组测序可用于研究微生物群体功能特性、相互协作关系及与环境之间的关系。与传统方法相比,无需纯培养,大大减少了研究的费用、周期并提升了物种丰富度。
发表期刊:Microbiome 影响因子:14.650 发表日期:2020年 发表单位:中山大学海洋科学学院
我们人类的肠道里寄宿着100~1500种近100万亿个细菌,在其他生物体内,这个数目也是非常可观的。这些细菌与宿主相互作用,正常情况下肠道内菌群间维持着共生或拮抗的关系,这是宿主肠道菌群能够处于微生态均衡的主要原因。越来越多的证据表明,一些复杂的疾病和宿主肠道微生物群(IM)的失调有很大关联。全肠道生态系统的改变,而不是单一的病原体,与白便综合征(WFS)有关,这是一种严重的全球性非传染虾病,但没有实验证据来探讨其因果关系。本研究通过全面的宏基因组和代谢组学分析,并进行了肠道微生物群移植(IMT),以研究IM失调与WFS之间的因果关系。
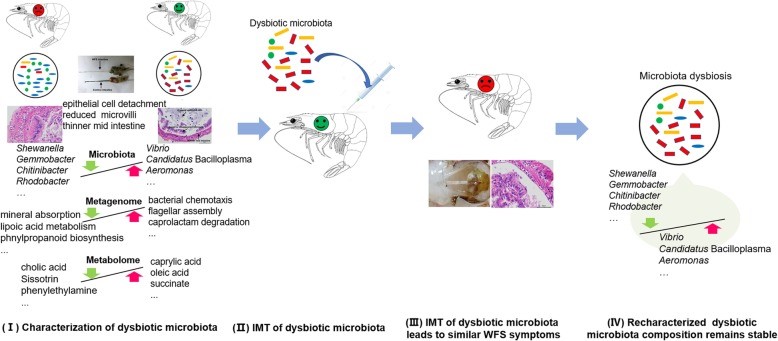
文章举例说明了IM失调在虾WFS发生中的致病作用。将研究设计和主要发现综合成一个概念模型,如图5所示。WFS虾的微生态科赫假设是(一)所有WFS虾中都具有类似的益生元IM;(二) 益生元IM可以回收并移植到健康虾身上;(三) 移植IM可导致与WFS虾相似的症状;(四)在新的WFS虾中,益生元IM成分的再特征化是一致的。
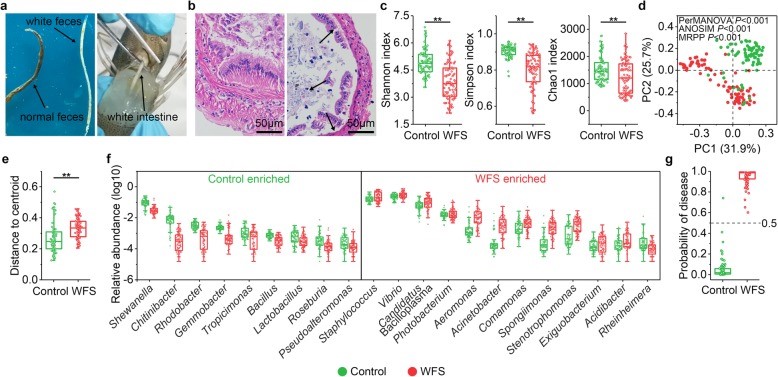 图1. WFS临床症状、组织病理学和微生物特征的表征
图1. WFS临床症状、组织病理学和微生物特征的表征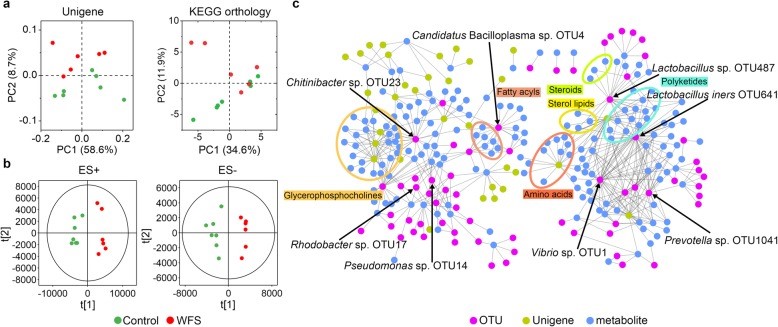 图2. 对照组和WFS之间微生物基因功能和代谢模式的比较分析
图2. 对照组和WFS之间微生物基因功能和代谢模式的比较分析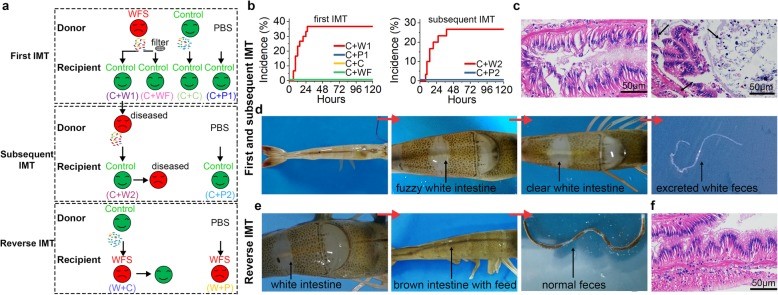 图3. IM移植导致类似的WFS症状,IMT程序的示意图
图3. IM移植导致类似的WFS症状,IMT程序的示意图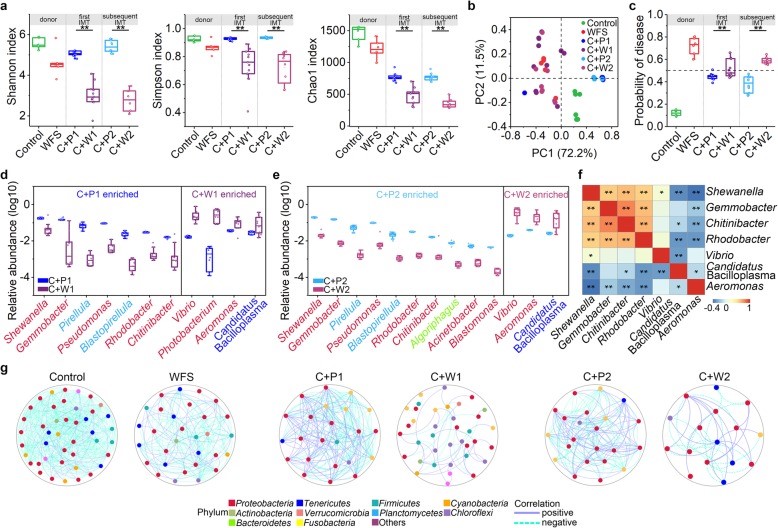 图4. 对新发病对虾的IM生态失调进行再特征描述
图4. 对新发病对虾的IM生态失调进行再特征描述这些原理通过多组学数据得到验证,揭示了IM失调与WFS发生之间的因果关系,从而从生态学角度对水生动物疾病病原学打开了新的认识。
Huang Z, Zeng S, Xiong J, Hou D, Zhou R, Xing C, Wei D, Deng X, Yu L, Wang H, Deng Z, Weng S, Kriengkrai S, Ning D, Zhou J, He J. Microecological Koch's postulates reveal that intestinal microbiota dysbiosis contributes to shrimp white feces syndrome. Microbiome. 2020 Mar 10;8(1):32. doi: 10.1186/s40168-020-00802-3. PMID: 32156316; PMCID: PMC7065354.
Copyright © 2015-2023 苏州帕诺米克生物医药科技有限公司 版权所有